









Inhalte
Tuberöse Sklerose nach Bourneville
Was ist es ?
Die tuberöse Sklerose (Bourneville) ist eine komplexe genetische Erkrankung, die durch die Entwicklung eines gutartigen (nicht krebsartigen) Tumors in verschiedenen Körperteilen gekennzeichnet ist. Diese Tumoren können dann in der Haut, im Gehirn, in den Nieren und anderen Organen und Geweben lokalisiert werden. Diese Pathologie kann auch ernsthafte Probleme in der Entwicklung des Individuums verursachen. Die klinischen Manifestationen und der Schweregrad der Erkrankung variieren jedoch von Patient zu Patient.
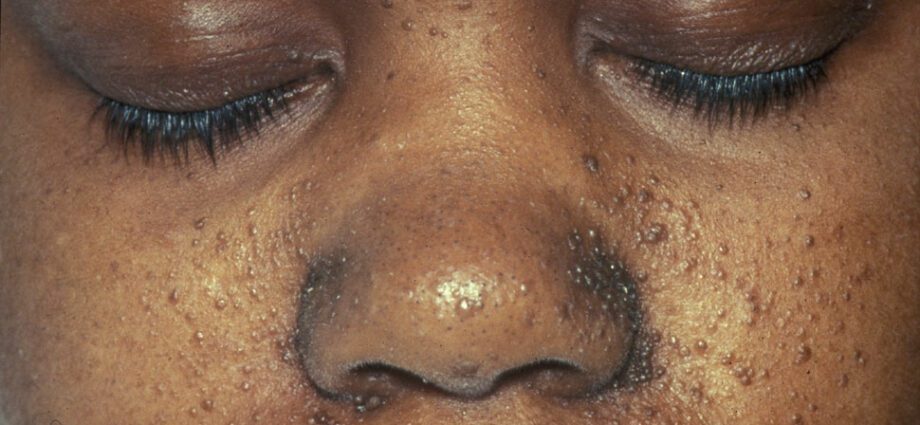
Die damit verbundenen Hautanomalien ähneln im Allgemeinen Flecken auf der Haut oder Bereichen, in denen die Haut heller ist als am Rest des Körpers. Die Entwicklung von Tumoren im Gesicht wird als Angiofibrom bezeichnet.
Im Zusammenhang mit einer Hirnschädigung sind die klinischen Symptome epileptische Anfälle, Verhaltensauffälligkeiten (Hyperaktivität, Aggressivität, geistige Behinderungen, Lernschwierigkeiten etc.). Einige Kinder mit der Krankheit haben sogar eine Form von Autismus, Entwicklungsstörungen, die soziale Interaktionen und Kommunikation beeinträchtigen. Gutartige Hirntumore können auch Komplikationen verursachen, die für das Subjekt tödlich sein können.
Die Entwicklung von Tumoren in den Nieren ist bei Menschen mit tuberöser Sklerose häufig. Dies kann zu schweren Komplikationen der Nierenfunktion führen. Darüber hinaus können sich Tumore im Herzen, in der Lunge und in der Netzhaut entwickeln. (2)
Es handelt sich um eine seltene Erkrankung, deren Prävalenz (Anzahl der Fälle in einer bestimmten Bevölkerung zu einem bestimmten Zeitpunkt) 1 / 8 bis 000 / 1 Personen beträgt. (fünfzehn)
Symptome
Die klinischen Manifestationen im Zusammenhang mit der tuberösen Sklerose von Bourneville variieren je nach den betroffenen Organen. Darüber hinaus variieren die mit der Krankheit verbundenen Symptome von Person zu Person stark. Mit leichten bis schweren Symptomen.
Die am häufigsten identifizierten Symptome dieser Krankheit sind epileptische Anfälle, kognitive und Verhaltensstörungen, Hautanomalien usw. Die am häufigsten betroffenen Organe sind: das Gehirn, das Herz, die Nieren, die Lunge und die Haut.
Die Entwicklung von bösartigen (Krebs-)Tumoren ist bei dieser Erkrankung möglich, jedoch selten und betrifft hauptsächlich die Nieren.
Die klinischen Anzeichen der Erkrankung im Gehirn stammen von Attacken auf verschiedenen Ebenen:
– Schäden an kortikalen Tuberkeln;
– Ependymknoten (SEN);
– Riesenependymale Astrozytome.
Sie führen zu: Entwicklung von geistiger Behinderung, Lernschwierigkeiten, Verhaltensstörungen, Aggressivität, Aufmerksamkeitsstörungen, Hyperaktivität, Zwangsstörungen usw.
Nierenschäden sind durch die Entwicklung von Zysten oder Angiomyolipomen gekennzeichnet. Diese können zu Nierenschmerzen und sogar zu Nierenversagen führen. Wenn starke Blutungen erkennbar sind, kann dies an einer schweren Anämie oder an Bluthochdruck liegen. Andere schwerwiegendere, aber seltene Folgen können ebenfalls sichtbar sein, insbesondere die Entwicklung von Karzinomen (Tumor der konstituierenden Zellen des Epithels).
Augenschäden können sichtbaren Flecken auf der Netzhaut ähneln und zu Sehstörungen oder sogar Erblindung führen.
Hautanomalien sind zahlreich:
– hypomelanische Makulae: die aufgrund eines Mangels an Melanin, einem Protein, das der Haut Farbe verleiht, zu hellen Flecken auf der Haut überall am Körper führen;
– das Auftreten von roten Flecken im Gesicht;
– verfärbte Stellen auf der Stirn;
– andere Hautanomalien, die von einer Person zur anderen abhängig sind.
Lungenläsionen sind bei 1/3 der Patienten mit einer leichten weiblichen Dominanz vorhanden. Die damit verbundenen Symptome sind dann mehr oder weniger starke Atembeschwerden.
Die Ursprünge der Krankheit
Der Ursprung der Krankheit ist genetisch und erblich.
Die Übertragung beinhaltet Mutationen in den TSC1- und TSC2-Genen. Diese interessanten Gene spielen bei der Bildung von Proteinen eine Rolle: Hamartin und Tuberin. Diese beiden Proteine ermöglichen es, durch ein interaktives Spiel die Zellproliferation zu regulieren.
Patienten mit der Krankheit werden mit mindestens einer mutierten Kopie dieser Gene in jeder ihrer Zellen geboren. Diese Mutationen begrenzen dann die Bildung von Hamartin oder Tubertin.
Wenn die beiden Kopien des Gens mutiert sind, verhindern sie die Produktion dieser beiden Proteine vollständig. Dieser Proteinmangel erlaubt es dem Körper daher nicht mehr, das Wachstum bestimmter Zellen zu regulieren und führt in diesem Sinne zur Entwicklung von Tumorzellen in verschiedenen Geweben und/oder Organen.
Risikofaktoren
Die Risikofaktoren für die Entwicklung einer solchen Pathologie sind genetisch bedingt.
Tatsächlich erfolgt die Übertragung der Krankheit durch einen autosomal-dominanten Modus. Entweder befindet sich das mutierte Gen von Interesse auf einem nicht-sexuellen Chromosom. Außerdem reicht das Vorhandensein nur einer der beiden Kopien des mutierten Gens aus, um die Krankheit zu entwickeln.
In diesem Sinne hat eine Person, die einen dieser beiden an der Krankheit erkrankten Elternteile besitzt, ein 50%iges Risiko, selbst den kranken Phänotyp zu entwickeln.
Prävention und Behandlung
Die Diagnose der Krankheit ist zunächst differentiell. Sie basiert auf atypischen physikalischen Kriterien. In den meisten Fällen sind die ersten charakteristischen Anzeichen der Krankheit: das Vorhandensein von wiederkehrenden epileptischen Anfällen und Verzögerungen in der Entwicklung des Patienten. In anderen Fällen führen diese ersten Anzeichen zu Hautflecken oder zur Erkennung eines Herztumors.
Nach dieser Erstdiagnose sind weitere Untersuchungen unumgänglich, um die Diagnose zu validieren oder nicht. Diese beinhalten:
– ein Gehirnscan;
– ein MRT (Magnetic Resonance Imaging) des Gehirns;
– ein Ultraschall des Herzens, der Leber und der Nieren.
Die Diagnose kann bei der Geburt des Kindes wirksam sein. Ansonsten ist es wichtig, dass es so schnell wie möglich durchgeführt wird, um den Patienten so schnell wie möglich zu betreuen.
Derzeit gibt es keine Heilung für die Krankheit. Die damit verbundenen Behandlungen sind daher unabhängig von den Symptomen jedes Einzelnen.
In der Regel werden Antiepileptika verabreicht, um die Anfälle zu begrenzen. Darüber hinaus werden auch Medikamente zur Behandlung von Tumorzellen des Gehirns und der Nieren verschrieben. Bei Verhaltensauffälligkeiten ist eine gezielte Behandlung des Kindes erforderlich.
Die Behandlung der Krankheit ist in der Regel langfristig. (1)