









Inhalte
Ewing-Sarkom
Was ist es ?
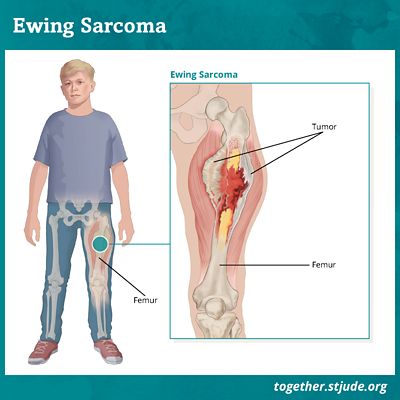
Das Ewing-Sarkom ist durch die Entwicklung eines bösartigen Tumors in den Knochen und Weichteilen gekennzeichnet. Dieser Tumor hat die Eigenschaft, ein hohes Metastasierungspotential zu haben. Entweder wird bei dieser Pathologie oft die Ausbreitung von Tumorzellen im ganzen Körper festgestellt.
Es ist eine seltene Krankheit, die im Allgemeinen Kinder betrifft. Die Inzidenz beträgt 1/312 Kinder unter 500 Jahren.
Die am stärksten von der Entwicklung dieser Tumorform betroffene Altersgruppe liegt zwischen 5 und 30 Jahren, mit einer noch höheren Inzidenz zwischen 12 und 18 Jahren. (3)
Die damit verbundenen klinischen Manifestationen sind Schmerzen und Schwellungen am Ort des Tumors.
Die für das Ewing-Sarkom charakteristischen Tumorzellen sind vielfältig: Beine, Arme, Füße, Hände, Brustkorb, Becken, Schädel, Wirbelsäule usw.
Dieses Ewing-Sarkom wird auch als primärer peripherer neuroektodermaler Tumor bezeichnet. (1)
Ärztliche Untersuchungen ermöglichen die mögliche Diagnose der Krankheit und bestimmen ihren Fortschritt. Die am häufigsten damit verbundene Untersuchung ist eine Biopsie.
Spezifische Faktoren und Bedingungen können die Prognose der Krankheit bei einem betroffenen Subjekt beeinflussen. (1)
Zu diesen Faktoren zählen insbesondere die Ausbreitung von Tumorzellen nur in die Lunge, deren Prognose günstiger ist, oder die Entwicklung von Metastasen in andere Körperteile. Im letzteren Fall ist die Prognose schlechter.
Darüber hinaus spielen die Größe des Tumors und das Alter des Betroffenen eine wesentliche Rolle für die Vitalprognose. In der Tat ist die Prognose besorgniserregender, wenn die Größe des Tumors auf mehr als 8 cm ansteigt. Was das Alter angeht, je früher die Diagnose der Pathologie gestellt wird, desto besser ist die Prognose für den Patienten. (4)
Das Ewing-Sarkom ist neben dem Chondrosarkom und dem Osteosarkom eine der drei Hauptformen des primären Knochenkrebses. (2)
Symptome
Die am häufigsten mit dem Ewing-Sarkom verbundenen Symptome sind sichtbare Schmerzen und Schwellungen in den betroffenen Knochen und Weichteilen.
Die folgenden klinischen Manifestationen können ihren Ursprung in der Entwicklung eines solchen Sarkoms haben: (1)
- Schmerzen und/oder Schwellungen in Armen, Beinen, Brust, Rücken oder Becken;
- das Vorhandensein von „Beulen“ an denselben Körperteilen;
- das Vorhandensein von Fieber ohne besonderen Grund;
- Knochenbrüche ohne Grund.
Die damit verbundenen Symptome hängen jedoch von der Lokalisation des Tumors sowie seiner Bedeutung für die Entwicklung ab.
Die Schmerzen, die der Patient mit dieser Pathologie empfindet, verstärken sich normalerweise im Laufe der Zeit.
Andere, weniger häufige Symptome können ebenfalls sichtbar sein, wie zum Beispiel: (2)
- ein hohes und anhaltendes Fieber;
- Muskelsteifheit;
- erheblicher Gewichtsverlust.
Ein Patient mit Ewing-Sarkom kann jedoch keine Symptome haben. In diesem Sinne kann der Tumor dann ohne besondere klinische Manifestation wachsen und so den Knochen oder die Weichteile befallen, ohne sichtbar zu sein. Im letzteren Fall ist die Frakturgefahr umso wichtiger. (2)
Die Ursprünge der Krankheit
Da es sich beim Ewing-Sarkom um eine Krebsart handelt, ist über den genauen Ursprung seiner Entstehung wenig bekannt.
Dennoch wurde eine Hypothese über die Ursache seiner Entwicklung aufgestellt. Tatsächlich betrifft das Ewing-Sarkom besonders Kinder über 5 Jahre und Jugendliche. In diesem Sinne wurde die Möglichkeit eines Zusammenhangs zwischen dem schnellen Knochenwachstum bei dieser Personengruppe und der Entwicklung des Ewing-Sarkoms angesprochen.
Die Pubertät bei Kindern und Jugendlichen macht Knochen und Weichteile anfälliger für die Entwicklung eines Tumors.
Die Forschung hat auch gezeigt, dass ein Kind, das mit einem Nabelbruch geboren wurde, dreimal häufiger ein Ewing-Sarkom entwickelt. (2)
Über diese oben genannten Hypothesen hinaus wurde auch der Ursprung des Vorliegens einer genetischen Translokation angeführt. An dieser Translokation ist das EWSRI-Gen (22q12.2) beteiligt. Eine t (11; 22) (q24; q12) Translokation innerhalb dieses interessierenden Gens wurde in fast 90 % der Tumoren gefunden. Darüber hinaus wurden viele genetische Varianten wissenschaftlich untersucht, an denen die Gene ERG, ETV1, FLI1 und NR4A3 beteiligt sind. (3)
Risikofaktoren
Aus der Sicht, wo die genauen Ursprünge der Pathologie bis heute noch wenig bekannt sind, sind es auch die Risikofaktoren.
Darüber hinaus würde ein Kind, das mit einem Nabelbruch geboren wurde, nach den Ergebnissen wissenschaftlicher Studien dreimal häufiger an Krebs erkranken.
Darüber hinaus können auf genetischer Ebene Translokationen innerhalb des EWSRI-Gens (22q12.2) oder genetische Varianten in den Genen ERG, ETV1, FLI1 und NR4A3 zusätzliche Risikofaktoren für die Entwicklung der Krankheit darstellen. .
Prävention und Behandlung
Die Diagnose des Ewing-Sarkoms basiert auf einer Differentialdiagnose durch das Vorliegen charakteristischer Symptome beim Patienten.
Nach der ärztlichen Analyse der schmerzenden und geschwollenen Stellen wird in der Regel eine Röntgenaufnahme verordnet. Andere medizinische Bildgebungssysteme können ebenfalls verwendet werden, wie zum Beispiel: Magnetic Reasoning Imaging (MRI) oder sogar Scans.
Eine Knochenbiopsie kann auch durchgeführt werden, um die Diagnose zu bestätigen oder nicht. Dazu wird eine Knochenmarkprobe entnommen und unter dem Mikroskop analysiert. Diese diagnostischen Techniken können nach Vollnarkose oder Lokalanästhesie durchgeführt werden.
Die Diagnose der Erkrankung muss so schnell wie möglich erfolgen, damit die Behandlung schnell erfolgt und somit die Prognose besser ist.
Die Behandlung des Ewing-Sarkoms ähnelt der allgemeinen Behandlung anderer Krebsarten: (2)
- Eine Operation ist ein wirksames Mittel, um diese Art von Sarkom zu behandeln. Der chirurgische Eingriff hängt jedoch von der Größe des Tumors, seiner Lage sowie seiner Ausbreitung ab. Ziel der Operation ist es, den durch den Tumor geschädigten Teil des Knochen- oder Weichgewebes zu ersetzen. Dazu kann eine Metallprothese oder ein Knochentransplantat zum Ersatz des betroffenen Bereichs verwendet werden. In extremen Fällen ist manchmal eine Amputation von Gliedmaßen erforderlich, um einen Krebsrückfall zu verhindern;
- Chemotherapie, die normalerweise nach einer Operation verwendet wird, um den Tumor zu verkleinern und die Heilung zu erleichtern.
- Strahlentherapie, wird auch häufig nach einer Chemotherapie, vor oder nach einer Operation eingesetzt, um die Größe des Tumors zu reduzieren und das Risiko eines Rückfalls zu vermeiden.