









Inhalte
Vaskulitis kleiner Kapillaren
Vaskulitis kleiner Kapillaren
Dies ist eine große Gruppe von Vaskulitiden der Wand von Arteriolen, Venolen oder Kapillaren, deren Prognose sehr variabel ist, je nachdem, ob es sich um eine reine oder systemische kutane Vaskulitis handelt.
Der häufigste klinische Aspekt ist die Purpura (violette Flecken, die beim Drücken nicht verblassen), die sich vor allem in den unteren Gliedmaßen vorwölbt und infiltriert, durch Stehen verschlimmert wird und verschiedene Formen annehmen kann (petechial und ekchymotisch, nekrotisch, pustelförmig…) oder Livedo, an den Beinen eine Art violettes Netz (Livedo reticularis) oder Sprenkelung (Livedo racemosa) bilden. Wir können auch ein Raynaud-Phänomen beobachten (einige Finger werden in der Kälte weiß).
Purpura und Livedo können mit anderen Hautläsionen (Papeln, Knötchen, nekrotische Läsionen, blutende Bläschen), fixierter Urtikaria, die nicht juckt, einhergehen.
Das Vorhandensein von Manifestationen außerhalb der Haut stellt einen Schwerkraftfaktor dar, der das Vorhandensein einer Gefäßbeteiligung in den Organen anzeigt:
- Gelenkschmerzen,
- Bauchschmerzen, schwarzer Stuhl, Transitstörungen,
- periphere Neuropathie
- Ödeme der unteren Gliedmaßen,
- Hyper-Blutdruck,
- Atembeschwerden, Asthma, Bluthusten …
Der Arzt verordnet Untersuchungen zur Suche nach Ursache und Schwere: Bluttest mit Blutbild, Entzündungssuche, Leber- und Nierentests etc. Lungenröntgen bei Atembeschwerden etc.).
Vaskulitis durch Infektion ausgelöst:
- bakteriell: Streptokokken, gramnegative Kokken (Gonokokken und Meningokokken)
- viral: Hepatitis, infektiöse Mononukleose, HIV usw.
- parasitär: Malaria …
- Pilz: Candida albicans …
Vaskulitis im Zusammenhang mit immunologischen Anomalien
- Typ II (gemischt monoklonal) und III (gemischt polyklonal) Kryoglobulinämie, assoziiert mit Autoimmunerkrankung, Infektion (insbesondere Hepatitis C) oder Blutkrankheit
- Hypocomplementémie (Mac Duffie Urticarienne Vaskularit)
- Hyperglobulinémie (Waldenströms Hyperglobulinémique Purple)
- Konnektivitis: Lupus, Gougerot-Sjögren-Syndrom, rheumatoide Arthritis …
- Vaskulitis von Blutkrankheiten und Malignomen
- Leukämie, Lymphom, Myelom, Krebs
- Vaskulitis im Zusammenhang mit ANCA (anti-neutrophile zytoplasmatische Antikörper)
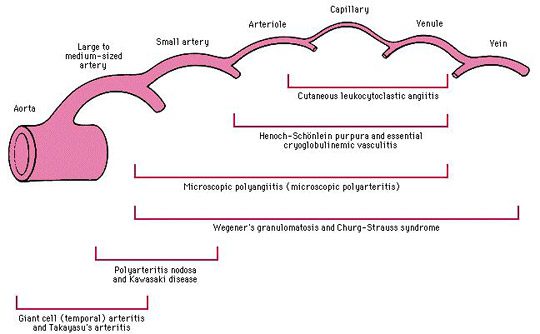
Micro Poly Angéite oder MPA
Die Mikropolyangiitis (MPA) ist eine systemische nekrotisierende Angeitis, deren klinische Symptome denen der PAN sehr ähnlich sind.
MPA ist mit ANCA vom Anti-Myeloperoxidase (Anti-MPO)-Typ assoziiert und führt typischerweise zu einer schnell fortschreitenden Glomerulonephritis und Lungenbeteiligung, die bei PAN nicht vorhanden ist.
Die Behandlung der MPA beginnt wie bei der PAN mit einer Kortikosteroidtherapie, manchmal kombiniert mit Immunsuppressiva (insbesondere Cyclophosphamid)
Morbus Wegener
Die Wegener-Granulomatose ist eine Vaskulitis, deren Beginn im Allgemeinen durch HNO- oder Atemwegssymptome (Sinusitis, Pneumopathie usw.) gekennzeichnet ist, die gegen Antibiotikabehandlungen resistent sind.
Klassischerweise führt eine diffuse Beteiligung der HNO (destruktive Pansinusitis), der Lunge (parenchymale Knötchen) und der renalen (halbmondförmige pauci-immune nekrotisierende Glomerulonephritis) zu der klassischen Trias der Wegener-Granulomatose.
Die Haut-Schleimhaut betrifft etwa 50% der Patienten: Purpura (violette Flecken, die beim Drücken nicht verschwinden) prall und infiltriert, Papeln, subkutane Knötchen, Hautulzerationen, Pusteln, Bläschen, hyperplastische Gingivitis …
ANCA ist ein diagnostischer und evolutionärer Test für Wegener-Granulomatose, mit diffuser zytoplasmatischer Fluoreszenz (c-ANCA), feinkörnig mit perinukleärer Anreicherung und / oder rein perinukleärer Fluoreszenz (p-ANCA).
Die Behandlung der Wegener-Granulomatose, die manchmal als medizinischer Notfall angesehen werden kann, sollte in einem spezialisierten Krankenhaus durch eine Kombination von Kortison und oralem Cyclophosphamid erfolgen.
Churg- und Strauss-Krankheit
Asthma ist ein wichtiges und frühes Kriterium dieser Vaskulitis, dem es im Durchschnitt 8 Jahre vor den ersten Anzeichen einer Vaskulitis (Neuropathie, Nebenhöhlenerkrankungen etc.) vorangeht und danach persistiert.
Blutuntersuchungen zeigen insbesondere einen deutlichen Anstieg der eosinophilen polynuklearen weißen Blutkörperchen
Die Behandlung des Morbus Churg und Strauss beginnt mit einer Kortikosteroidtherapie, manchmal kombiniert mit Immunsuppressiva (insbesondere Cyclophosphamid).
Meinung unseres Arztes
Infiltrierte Purpura (violette, etwas dicke Flecken, die nicht durch Fingerdruck verblassen) ist das Hauptzeichen einer Vaskulitis. Leider ist dieses Zeichen nicht immer vorhanden und die Variabilität unspezifischer klinischer Symptome erschwert Ärzten oft die Diagnose. Ebenso ist es bei der Kleingefäßvaskulitis, die im Vergleich zur Mittel- und Großgefäßvaskulitis mit Abstand der wichtigste Fall ist, der in der gegenwärtigen Praxis auftritt, bei weitem der wichtigste Fall, oft schwierig, eine zu behandelnde Ursache zu finden: etwa die Hälfte der Kleingefäßvaskulitis. Gefäße haben bei den biologischen und radiologischen Untersuchungen, die der Arzt zur Suche nach einer Ätiologie durchführt, keine Ursache gefunden. Wir sprechen oft von „allergischer Vaskulitis“ oder „Hypersensitivitätsvaskulitis“ bzw. „kutaner Vaskulitis kleiner Gefäße idiopathischen Kalibers“. Dr. Ludovic Rousseau, Dermatologe |
Sehenswürdigkeiten
Französische Studiengruppe Vaskulitis: www.vascularites.org
Dermatonet.com, Informationsseite zu Haut, Haar und Schönheit von einem Dermatologen
MedicineNet: http://www.medicinenet.com/vasculitis/article.htm