









Inhalte
Hirschsprung-Krankheit
Was ist es ?
Die Hirschsprung-Krankheit (HSCR) ist durch eine Lähmung im terminalen Teil des Dickdarms gekennzeichnet.
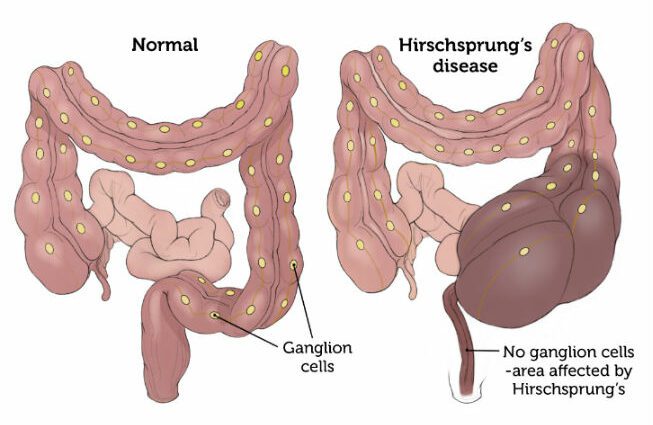
Diese Pathologie tritt von Geburt an auf und ist die Folge des Fehlens von Nervenganglien (Zellen, die auf dem Weg des Nervs eine Ausbuchtung bilden) in der Darmwand.
Das Schlucken von Nahrung über den Verdauungstrakt bis zur Ausscheidung ist zu einem großen Teil dank der Darmperistaltik möglich. Diese Peristaltik ist eine Reihe von Kontraktionen der Darmmuskulatur, die das Vordringen des Nahrungsbolus entlang des Verdauungstraktes ermöglichen.
In dieser Situation, in der im Dickdarm keine Nervenganglien vorhanden sind, wird die Peristaltik vom Körper nicht mehr bereitgestellt. In diesem Sinne entsteht eine Erweiterung des Darms und eine Volumenzunahme.
Die damit verbundenen Symptome sind umso wichtiger, wenn der Bereich der Nervenganglien groß ist. (1)
Diese Krankheit wird daher durch atypische Darmsymptome definiert: Darmverschluss. Es ist eine Blockade von Transit und Gas, die zu Bauchschmerzen, Koliken (Darmkrämpfen), Übelkeit, Blähungen usw. führt.
HSCR betrifft etwa 1 von 5 Geburten pro Jahr. Die Form, die den terminalen Teil des Dickdarms (Dickdarm) betrifft, betrifft hauptsächlich Jungen. (000) Mädchen sind anfälliger für die Entwicklung dieser Krankheit in einer weiter verbreiteten Form. (2)
Diese Pathologie betrifft hauptsächlich Babys und Kleinkinder. (3)
Mehrere Formen der Krankheit wurden nachgewiesen (2):
– die „klassische“ Form oder auch „Kurzsegmentform“ genannt. Diese Form ist bei Patienten mit dieser Pathologie am häufigsten, bis zu 80%. Diese Form der Krankheit betrifft den terminalen Teil des Dickdarms bis zum Rektumsegment;
– die „langsegmentige“ Form, die sich bis zum Sigma erstreckt, betrifft fast 15 % der Patienten;
– die Form der „totalen Kolik“, die den gesamten Dickdarm betrifft, betrifft 5 % der Patienten.
Symptome
Die Darmpassage wird vom Nervensystem gesteuert. Nervenganglien befinden sich daher im Darm und ermöglichen die Übertragung von Informationen aus dem Gehirn zur Steuerung der Darmperistaltik und damit des Nahrungsverlaufs entlang des Verdauungstraktes.
Ein Fehlen dieser Knoten verhindert bei Morbus Hirschsprung jegliche Informationsübertragung und blockiert somit die Darmperistaltik. Nahrung kann den Darm nicht mehr passieren und wird im Verdauungstrakt blockiert.
Die Symptome dieser Krankheit machen sich in der Regel sehr früh bei der Geburt bemerkbar. In einigen Fällen können sie jedoch nach ein oder zwei Jahren auftreten. (3)
Symptome, die Neugeborene und Kinder betreffen, sind hauptsächlich:
– Transitschwierigkeiten;
– Unfähigkeit, Mekonium (erste Exkremente des Neugeborenen) während der ersten 48 Stunden auszuscheiden;
- Verstopfung;
– Gelbsucht;
– Erbrechen;
- Durchfall;
– Bauchschmerzen;
– Unterernährung.
Symptome bei älteren Kindern sind:
– schwere Verstopfung mit Komplikationen (Gedeihstörung in Größe und Gewicht);
– schlechte Ernährung;
- Blähungen;
- ein Fieber.
Das Kind kann auch Darminfektionen wie Enterokolitis entwickeln.
Weitere Anomalien können ebenfalls sichtbar sein: Innenohrschwerhörigkeit (Waardenburg-Shah-Syndrom), geistige Behinderung (Mowat-Wilson-Syndrom), zentrale alveoläre Hypoventilation (Haddad-Syndrom), Gliedmaßenanomalien (Bardet-Syndrom) Biedl), medullärer Schilddrüsenkrebs (multiple endokrine .) Neoplasie Typ 2B) oder Chromosomenanomalien (Down-Syndrom). (2)
Die Ursprünge der Krankheit
Die Hirschsprung-Krankheit wird durch eine Anomalie in der Entwicklung des enterischen Nervensystems verursacht. Es handelt sich um eine Aganglionose, dh das Fehlen von Nervenganglien (auch „Cajalzellen“ genannt) im Darm. Dieses Lymphknotendefizit ist insbesondere im Endbereich des Dickdarms (Kolon) lokalisiert.
Bei der Person, die von dieser Pathologie betroffen ist, verbleibt dieser Teil des Darms daher in einem Zustand tonischer und dauerhafter Kontraktion. Diese Situation führt zu einem Darmverschluss. (2)
Sowohl genetische als auch umweltbedingte Faktoren sind an der Entstehung der Hirschsprung-Krankheit beteiligt. (2)
Tatsächlich wurden bestimmte Gene bei der Entwicklung dieser Pathogenese nachgewiesen. Es handelt sich um eine polygenetische Erkrankung, die insbesondere die Gene betrifft:
- Proko-Onkogen ret (RET);
– das aus Gliazellen stammende Gen für den neutrotrophen Faktor (GDNF);
– das Endothelin-Rezeptor-Gen vom Typ B (EDNRB);
– das Endothelin-3-Gen (EDN3);
– das Gen für das Endothelin 1 Converting Enzym 1 (ECE1);
– das Gen für das Zelladhäsionsmolekül L1 (L1CAM).
Risikofaktoren
Wie bereits erwähnt, ist die Hirschsprung-Krankheit die Folge des Fehlens von Nervenganglien im Dickdarm bis zum After, die die Darmperistaltik und damit das Aufsteigen der Nahrung auf diese Ebene verhindern.
Dieses Defizit an Cajal-Zellen (Nervenganglien) ist die Folge eines Wachstumsdefizits dieser Zellen während der fetalen Entwicklung. Die Ursachen für dieses fehlende Zellwachstum vor der Geburt sind noch nicht bekannt. Dennoch wurde die Möglichkeit eines Zusammenhangs zwischen dem allgemeinen Gesundheitszustand der Mutter während ihrer Schwangerschaft und dem Fehlen dieser Art von Zellen im Fötus vorgeschlagen.
Viele Gene wurden bei der Entwicklung der Krankheit nachgewiesen. Das Vorhandensein dieser Gene kann innerhalb derselben Familie häufig vorkommen. Ein Teil der Vererbung wäre dann der Ursprung der Entwicklung dieser Krankheit.
Darüber hinaus können auch bestimmte Pathologien ein zusätzlicher Risikofaktor für die Entwicklung des Morbus Hirschsprung sein. Dies ist insbesondere beim Down-Syndrom der Fall. (3)
Prävention und Behandlung
Die Differenzialdiagnose erfolgt nach den charakteristischen Symptomen der vom Patienten dargestellten Erkrankung: Darmverschluss, anorektale Stenose, Beckentumore usw. (2)
Die am häufigsten mit der Krankheit verbundene Diagnose wird durch eine rektale Biopsie gestellt. Diese Biopsie zeigt das Vorhandensein oder Fehlen von Nervenganglien im Dickdarm. Außerdem eine Überexpression von Acetylcholinesterase (Enzym, das die Hydrolyse von Acetylcholin zu Essigsäure und Cholin ermöglicht). (2)
Bei der Diagnose dieser Pathologie kann auch ein Bariumeinlauf (Röntgenuntersuchung zur Darstellung des Dickdarms) durchgeführt werden. Diese Methode ermöglicht es, einen vorübergehenden Bereich des Fehlens von Nervenzellen zu visualisieren, der auf die Entwicklung der Hischsprung-Krankheit hinweist. Dieses Diagnoseverfahren ist jedoch nicht 100% zuverlässig. Tatsächlich würden 10 bis 15 % der Fälle von Morbus Hirschsprung nach diesem Diagnoseversuch nicht diagnostiziert werden. (4)
Die wichtigste Behandlung der Krankheit ist eine Operation. Es ermöglicht die Ablation des Teils des Darms, der an Nervenzellen fehlt. (4)
Bei einer Totalschädigung des Dickdarms kann eine Dickdarmtransplantation erforderlich sein. (2)
Anschließend kann ein Stoma (chirurgische Technik, die die Verbindung zweier Organe ermöglicht) durchgeführt werden, um den operierten Darmabschnitt mit dem After oder mit dem oberen Darmabschnitt zu verbinden. Dieses Stoma kann je nach Fall entweder dauerhaft oder vorübergehend sein. (4)
Eine Operation hilft, die mit der Krankheit verbundenen Symptome zu reduzieren. Die Prognose ist jedoch nicht vollständig und entzündliche Komplikationen können auftreten und tödlich sein.